-
Antenna Effect in Noble Metal-Free Dye-Sensitized Photocatalytic Systems Enhances CO2-to-CO Conversion
V. Nikolaou, C. Govind, E. Balanikas, J. Bharti, S. Diring, E. Vauthey, M. Robert and F. Odobel
Angewandte Chemie International Edition, 63 (2024), p202318299


DOI:10.1002/anie.202318299 | unige:175842 | Abstract | Article HTML | Article PDF | Supporting Info
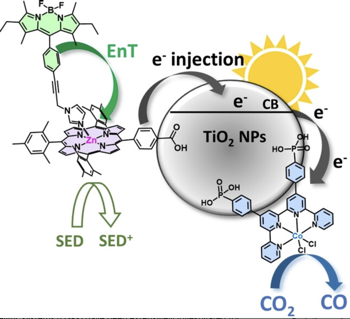
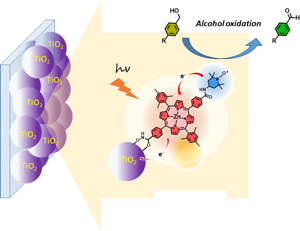
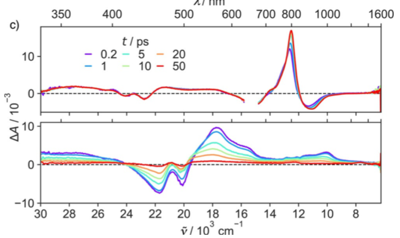
Dye-sensitized photocatalytic systems (DSPs) have been extensively investigated for solar-driven hydrogen (H2) evolution. However, their application in carbon dioxide (CO2) reduction remains limited. Furthermore, current solar-driven CO2-to-CO DSPs typically employ rhenium complexes as catalysts. In this study, we have developed DSPs that incorporate noble metal-free components, specifically a zinc-porphyrin as photosensitizer (PS) and a cobalt-quaterpyridine as catalyst (CAT). Taking a significant stride forward, we have achieved an antenna effect for the first time in CO2-to-CO DSPs by introducing a Bodipy as an additional chromophore to enhance light harvesting efficiency. The energy transfer from Bodipy to zinc porphyrin resulted in remarkable stability (turn over number (TON)=759 vs. CAT), and high CO evolution activity (42?mmol?g?1?h?1 vs. CAT).